EN ISO 13485 is the internationally recognized standard for quality management systems in the design and manufacture of medical devices. For companies selling medical devices to enterprise clients, this certification represents far more than regulatory housekeeping. It outlines specific requirements that help organizations ensure their medical devices meet both customer and regulatory demands for safety and efficacy.
Enterprise procurement teams expect demonstrable quality management frameworks before qualifying suppliers for long-term contracts. EN ISO 13485 certification provides systematic evidence that your organization maintains controlled design processes, validated manufacturing operations, and documented risk management throughout the product lifecycle. This standard not only facilitates market access across different countries but also enhances trust among stakeholders through demonstrated commitment to safety and quality.
The "EN" prefix indicates adoption by European standards bodies, making ISO 13485 formally recognized within EU regulatory frameworks. This alignment proves particularly significant for organizations pursuing CE marking under the Medical Device Regulation (MDR) or In Vitro Diagnostic Regulation (IVDR).
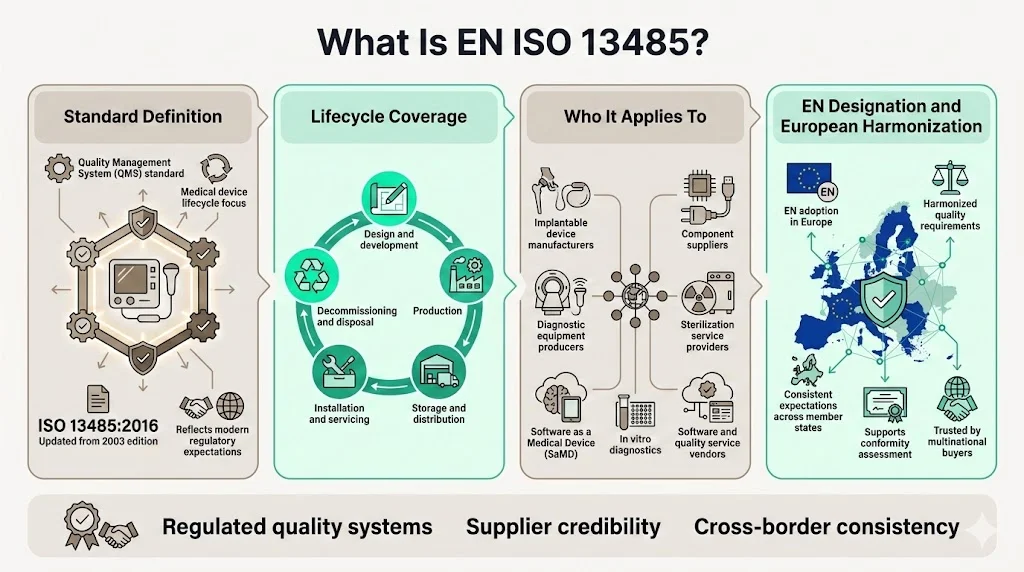
What Is EN ISO 13485?
ISO 13485 specifies requirements for a quality management system that can be used by an organization involved in one or more stages of the life-cycle of a medical device, including design and development, production, storage and distribution, installation, servicing and final decommissioning and disposal of medical devices. The current version, ISO 13485:2016, replaced the 2003 edition following significant evolution in regulatory expectations and medical device complexity.
The standard applies to manufacturers of implantable devices, diagnostic equipment, software as a medical device (SaMD), in vitro diagnostics, and related services. It can also benefit suppliers and external parties that provide products, including quality management system-related services to such organizations. Organizations producing components, sterilization services, or software tools for regulated device manufacturers frequently implement ISO 13485 to meet customer contractual requirements.
European adoption through the EN designation ensures consistency across member states and provides a recognized foundation for conformity assessment. This harmonization matters substantially when enterprise buyers operate facilities across multiple jurisdictions—they expect unified quality standards from suppliers regardless of manufacturing location.
Building toward SOC 2, ISO 27001, or another framework?
Drop your work email and we'll help you turn security work into audit-ready evidence automatically.
Why EN ISO 13485 Matters for Enterprise-Focused Companies
Enterprise procurement in regulated industries operates fundamentally differently from transactional software or component purchasing. Large healthcare organizations, hospital systems, and medical device integrators conduct thorough supplier qualification processes before awarding contracts. ISO 13485 certification reduces procurement friction by providing third-party verification of quality management capabilities.
Vendor qualification audits from enterprise buyers examine design controls, manufacturing process validation, complaint handling procedures, and post-market surveillance systems. Organizations holding current ISO 13485 certification demonstrate these systems already meet internationally recognized standards, reducing buyer risk and shortening qualification timelines. This advantage proves decisive in competitive procurement scenarios where multiple technically qualified suppliers compete.
Contract terms increasingly specify ISO 13485 maintenance as a continuing requirement throughout the supplier relationship. Loss of certification typically triggers material breach clauses and termination rights. Organizations treating certification as a one-time achievement rather than sustained operational discipline face significant commercial consequences when surveillance audits identify major nonconformities.
Regulatory convergence around ISO 13485 intensifies its importance for global market access. The FDA amended 21 CFR 820 by incorporating by reference ISO 13485:2016, aligning U.S. quality system requirements with the international standard. Companies maintaining ISO 13485 conformance position themselves for streamlined regulatory submissions across multiple jurisdictions rather than managing fragmented quality system requirements.
Core Principles of EN ISO 13485
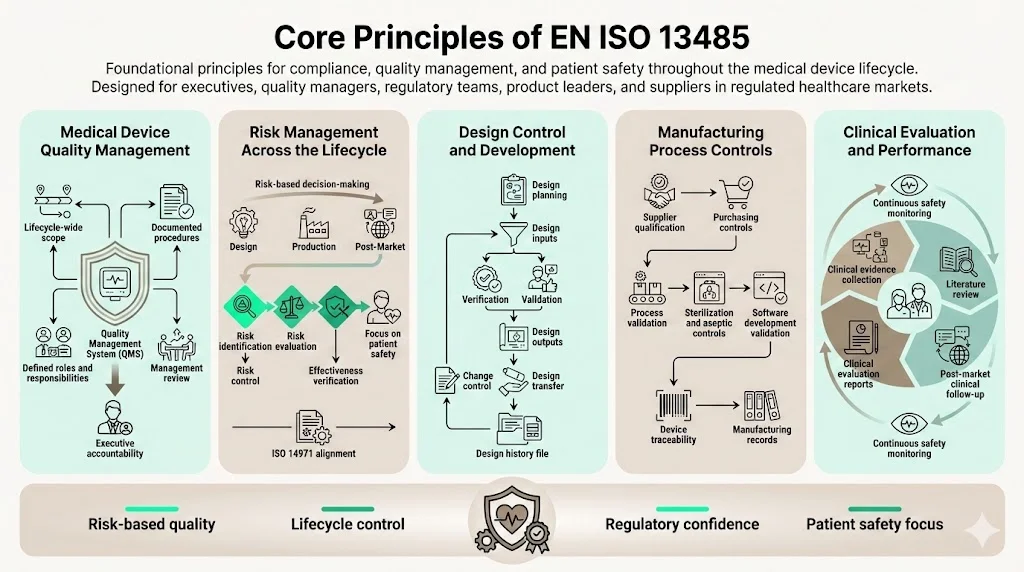
1) Medical Device Quality Management
ISO 13485 requires establishing a quality management system encompassing all lifecycle stages relevant to your organization's scope. This process-based approach extends beyond manufacturing to include design inputs, supplier management, customer feedback, and post-market activities. The standard mandates documented procedures, defined responsibilities, and management review processes ensuring quality objectives align with regulatory requirements.
Organizations must establish quality policies, objectives, and planning mechanisms that demonstrate top management commitment. This differs substantially from delegating compliance to quality departments—ISO 13485 requires management involvement in resource allocation, infrastructure provision, and strategic quality planning.
2) Risk Management Across the Product Lifecycle
ISO 13485:2016 has a greater emphasis on risk management and risk-based decision making, as well as changes related to the increased regulatory requirements for organizations in the supply chain. Risk management integration extends throughout design, purchasing, production, installation, and servicing activities. Organizations must implement systematic methods for identifying hazards, evaluating risks, implementing controls, and verifying effectiveness.
The standard requires documented risk management processes typically implemented through ISO 14971, the specific standard for medical device risk management. Risk assessments inform design decisions, manufacturing process controls, supplier evaluation criteria, and post-market surveillance planning. This risk-based approach ensures quality system resources focus where actual patient safety and product performance risks exist.
3) Design Control and Development
ISO 13485 establishes detailed design control requirements addressing planning, inputs, outputs, review, verification, validation, transfer, and change control. Design inputs must capture intended use, performance requirements, regulatory requirements, and applicable standards. Organizations must demonstrate systematic design verification confirming outputs meet inputs, and design validation proving devices meet user needs under actual conditions.
Design change control provisions require evaluating modifications for regulatory impact, implementing changes through controlled processes, and maintaining design history files documenting the complete development evolution. These requirements address the reality that inadequate design controls represent a leading cause of device failures, recalls, and regulatory enforcement actions.
4) Manufacturing Process Controls
The standard mandates process validation for production operations where results cannot be fully verified through subsequent inspection and testing. This encompasses sterilization processes, aseptic manufacturing, and software development. Organizations must establish validation protocols, document acceptance criteria, conduct process qualification studies, and implement ongoing process monitoring.
Supplier controls require organizations to evaluate and select suppliers based on their ability to meet requirements, implement purchasing specifications, and verify purchased product conformity. Traceability requirements mandate maintaining records connecting device identification through manufacturing history to distribution records—critical for investigating complaints, managing recalls, and demonstrating conformity during regulatory inspections.
5) Clinical Evaluation and Performance
ISO 13485 requires organizations to establish procedures for collecting and reviewing clinical data demonstrating device safety and performance. This includes clinical investigation planning, ongoing literature review, post-market clinical follow-up activities, and clinical evaluation report maintenance. The standard aligns with EU MDR clinical evidence requirements and regulatory authority expectations for data-driven safety assessments.
Clinical evaluation represents an ongoing lifecycle activity rather than a pre-market exercise. Organizations must continuously evaluate emerging clinical data, competitor information, literature findings, and post-market surveillance data to identify potential safety issues or performance concerns requiring corrective action.
EN ISO 13485 and Regulatory Compliance
EN ISO 13485 certification supports CE marking processes under EU MDR and IVDR by demonstrating conformance to quality system requirements. Notified Bodies conducting conformity assessments evaluate quality management system implementation as part of device approval. Organizations lacking ISO 13485 certification face substantially more intensive scrutiny during notified body audits.
The FDA has determined that the requirements in ISO 13485 are, when taken in totality, substantially similar to the requirements of the QS regulation, providing a similar level of assurance in a firm's quality management system. This regulatory convergence reduces compliance burden for organizations operating in multiple markets. Rather than maintaining separate quality systems for different jurisdictions, companies implement one ISO 13485-conformant system satisfying multiple regulatory authorities.
Regulatory inspections evaluate actual quality system implementation, not merely certification status. A certificate of conformance to ISO 13485 will not exempt a manufacturer from an FDA inspection. However, organizations maintaining robust ISO 13485 conformance typically perform better during regulatory inspections because their quality systems already address fundamental regulatory expectations around documentation, process control, and continuous improvement.
Quality Assurance and Documentation Requirements
ISO 13485 mandates specific documented procedures covering quality system management, document control, record control, internal audit, corrective and preventive action, and other quality processes. Organizations must establish documentation hierarchies typically including quality manuals, procedures, work instructions, and records demonstrating conformance.
Document control requirements address document approval, revision management, availability at points of use, and obsolete document removal. Changes to documents affecting product quality require review and approval before implementation. Electronic document management systems prove essential for organizations scaling operations or managing geographically distributed teams—paper-based systems cannot maintain the control, traceability, and accessibility ISO 13485 requires.
Nonconformity handling procedures must address product and quality system nonconformances through systematic investigation, root cause analysis, correction, corrective action, and effectiveness verification. Organizations frequently struggle distinguishing between correction (fixing the immediate problem) and corrective action (eliminating the root cause). Regulatory authorities specifically examine whether corrective actions address systemic issues or merely treat symptoms.
Auditing and Inspection Under EN ISO 13485
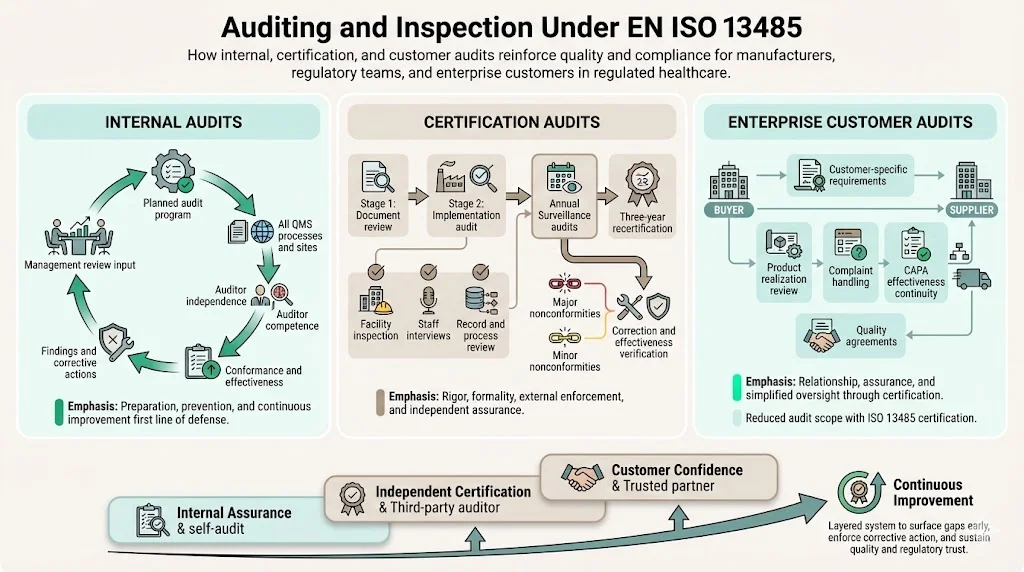
Internal Audits
ISO 13485 requires planned internal audits evaluating quality management system conformance, effectiveness, and maintenance. Organizations must establish audit programs covering all quality system processes and locations at planned intervals. Auditors must possess independence from the activities being audited and demonstrate competence in both auditing techniques and applicable regulatory requirements.
Internal audit findings drive management review and continuous improvement. Organizations treating internal audits as perfunctory exercises inevitably face more severe findings during external certification audits and regulatory inspections. Effective internal audit programs identify gaps, drive corrective actions, and verify improvement effectiveness before external assessments occur.
Certification Audits
Third-party certification follows a structured process beginning with Stage 1 document review evaluating quality system documentation against ISO 13485 requirements. Stage 2 implementation audits verify actual conformance through facility inspection, employee interviews, process observation, and record review. Auditors evaluate whether documented procedures reflect actual practices and whether the quality system achieves intended results.
Third-party audits assess conformance to a standard and occur annually based on a three-year plan. Following initial certification, surveillance audits occur at defined intervals—typically annually—verifying continued conformance, evaluating corrective actions from previous audits, and assessing quality system changes. Recertification audits occur every three years, providing comprehensive reassessment of the entire quality management system.
Major nonconformities identified during certification or surveillance audits can result in certification suspension or withdrawal. Organizations must implement effective corrective actions and demonstrate sustained improvement to maintain certification status.
Enterprise Customer Audits
Large medical device buyers and healthcare organizations frequently conduct supplier audits supplementing ISO 13485 certification. These audits examine specific processes relevant to the buyer relationship—product realization activities, complaint handling responsiveness, corrective action effectiveness, and supply chain continuity planning. Customer audit findings may impose requirements exceeding ISO 13485 baseline expectations.
ISO 13485 certification substantially reduces customer audit burden by providing standardized evidence of quality system conformance. Rather than explaining fundamental quality processes, certified organizations focus customer audits on product-specific controls, performance data, and relationship-specific quality agreements.
EN ISO 13485 vs Other ISO Standards
While ISO 9001 applies to a wide range of industries, ISO 13485 is specifically tailored to the regulatory and safety requirements of the medical device industry. It emphasizes meeting regulatory as well as customer requirements, risk management, and effective process validation more than ISO 9001.
ISO 9001 embraces flexible continual improvement principles allowing organizations substantial latitude in quality system design. ISO 13485 imposes prescriptive requirements reflecting regulatory expectations for medical device manufacturing. This includes mandatory documented procedures, specific validation requirements, sterile device provisions, and regulatory reporting obligations absent from ISO 9001.
Organizations holding ISO 9001 certification cannot assume compliance with ISO 13485. The medical device standard requires additional controls addressing design validation, process validation, traceability, sterile barrier systems, and clinical evaluation. Enterprise buyers in regulated industries specifically require ISO 13485—general quality management certifications prove insufficient for demonstrating medical device manufacturing competence.
Some organizations maintain both certifications, using ISO 9001 for non-medical product lines and ISO 13485 for regulated medical device operations. This dual approach requires careful scope definition, quality system segregation where necessary, and certification body coordination to avoid audit duplication.
Implementation Challenges and Practical Considerations
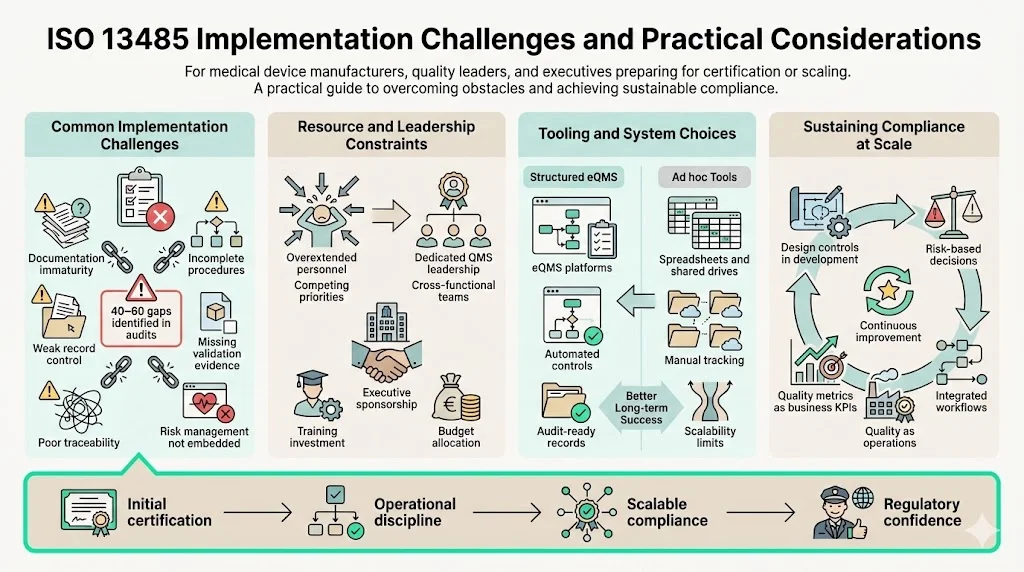
Organizations pursuing initial ISO 13485 certification commonly struggle with documentation maturity, process validation evidence, risk management integration, and management commitment beyond policy statements. Gap analyses conducted by experienced auditors typically identify 40-60 deficiencies requiring remediation before certification readiness.
Resource planning proves critical—organizations cannot successfully implement ISO 13485 by adding responsibilities to already-overextended staff. Successful implementations assign dedicated quality system leadership, engage cross-functional teams, and secure executive sponsorship including budget allocation for training, tooling, and external expertise.
Software selection significantly impacts implementation efficiency and long-term sustainability. Purpose-built electronic quality management systems (eQMS) designed for medical device regulatory requirements provide structured workflows, automated compliance controls, and audit-ready documentation. Organizations attempting ISO 13485 implementation using general document management systems, spreadsheets, or shared drives inevitably face scalability limitations as product complexity and regulatory scope expand.
Maintaining compliance as organizations scale requires embedding quality system thinking into operational culture rather than treating it as overhead. High-performing organizations integrate design controls into development workflows, incorporate risk management into decision processes, and position quality metrics as business performance indicators rather than compliance artifacts.
Business Impact of EN ISO 13485 Certification
ISO 13485 certification provides tangible competitive advantages in enterprise sales cycles. Procurement teams use certification status as initial supplier screening criteria—organizations lacking certification often face disqualification before technical evaluation. This gating function intensifies in competitive markets where multiple certified suppliers exist.
Regulatory submissions, notified body assessments, and authority inspections proceed more efficiently when organizations demonstrate ISO 13485 conformance. Rather than explaining basic quality system structures, interactions focus on product-specific evidence and regulatory questions. This efficiency reduces time to market and regulatory approval costs.
Risk management during post-market activities improves substantially under ISO 13485 frameworks. Systematic complaint handling, trend analysis, corrective action processes, and regulatory reporting procedures reduce liability exposure and support effective recall execution when necessary. Enterprise buyers specifically evaluate suppliers' post-market quality system maturity when making long-term strategic sourcing decisions.
Quality system maturity directly impacts product quality, manufacturing efficiency, and customer satisfaction. Organizations implementing ISO 13485 effectively report reduced rework, improved first-pass yield, faster issue resolution, and enhanced customer confidence. These operational benefits compound over time as quality data informs continuous improvement initiatives.
Conclusion
EN ISO 13485 represents the operational foundation for medical device manufacturers competing in enterprise markets and regulated jurisdictions. Organizations treating certification as a paper exercise satisfying auditors while maintaining separate "real" work processes inevitably face quality failures, regulatory enforcement, and enterprise customer defection. Effective implementation requires integrating quality system requirements into actual business operations, demonstrating management commitment through resource allocation, and maintaining continuous conformance rather than preparing for periodic audits.
The regulatory landscape continues evolving with increased clinical evidence expectations, heightened post-market surveillance requirements, and intensified supply chain scrutiny. Organizations establishing robust ISO 13485 conformance position themselves to adapt efficiently to regulatory changes while maintaining enterprise customer confidence. This strategic quality system investment yields returns through improved market access, reduced compliance costs, and sustained competitive differentiation in B2B medical device markets.
Frequently Asked Questions (FAQ)
1) Who needs EN ISO 13485 certification?
ISO 13485 is designed to be used by organizations involved in the design, production, installation and servicing of medical devices and related services. This encompasses device manufacturers, contract manufacturers, sterilization service providers, software developers creating SaMD, and component suppliers. Organizations serving multiple customers in regulated markets pursue certification to satisfy contractual requirements and reduce repetitive customer audits. Distributors and importers in certain jurisdictions also require certification depending on local regulatory frameworks and the device classification they handle.
2) How is EN ISO 13485 different from ISO 9001?
ISO 13485 provides medical device-specific requirements addressing regulatory compliance, risk management rigor, design control depth, process validation mandates, and sterile device provisions absent from ISO 9001. The medical device standard emphasizes regulatory requirement conformance alongside customer satisfaction, while ISO 9001 prioritizes customer focus and continual improvement flexibility. ISO 13485 requires specific documented procedures and imposes prescriptive controls reflecting medical device regulatory expectations. Organizations certified to ISO 9001 cannot assume ISO 13485 conformance without implementing additional medical device-specific requirements.
3) Does EN ISO 13485 apply outside the EU?
The EN prefix indicates European adoption, but ISO 13485 serves as the globally recognized medical device quality standard. Regulatory authorities in Canada, Australia, Japan, Brazil, and numerous other jurisdictions recognize ISO 13485 as evidence of quality system conformance. The FDA's integration of ISO 13485:2016 into 21 CFR 820 extends this recognition to the United States market. Enterprise medical device buyers worldwide specify ISO 13485 certification regardless of geographic location, making it the de facto international quality system standard for medical device supply chains.
4) How long does certification take?
Organizations beginning with minimal quality system maturity typically require 9-12 months for ISO 13485 implementation before certification readiness. This timeline encompasses gap analysis, procedure development, training, process implementation, internal auditing, and management review cycles necessary to demonstrate sustained conformance. Companies with established quality foundations or ISO 9001 certification often achieve certification in 6-8 months. Certification body scheduling, organizational complexity, and product portfolio scope influence actual timelines. Organizations rushing implementation to meet artificial deadlines inevitably face certification delays when auditors identify fundamental conformance gaps requiring extensive corrective action.
5) What audits are required?
ISO 13485 certification requires internal audits covering all quality system processes before initial certification and periodically thereafter—typically annually at minimum. Certification bodies conduct Stage 1 documentation review followed by Stage 2 implementation audit before granting initial certification. Following certification, surveillance audits occur annually or semi-annually depending on certification body practices and organizational risk factors. Comprehensive recertification audits occur every three years, reassessing the entire quality management system. Organizations also face regulatory inspections from authorities like the FDA, Health Canada, or competent authorities in EU member states independent of certification audit schedules. Enterprise customers may conduct additional supplier audits based on contractual terms and relationship significance.
